

Editor’s Note: This story, first published in 2004, has been updated.
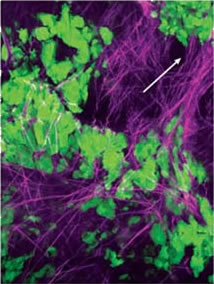
When John Condeelis, Ph.D., first watched tumor cells in living tissue under the microscope, he was amazed by what he saw. The cells were speeding along like little cars on fibrous “superhighways.” Their destination: a newly formed blood vessel surrounded by macrophages.
Macrophages are a type of white blood cell that normally are involved in inflammation and fighting infection. In this case, they appeared to be attracting the tumor cells to the vessel. “It’s almost as if the macrophages are sending a ‘come hither’ signal,” says his colleague Jeffrey W. Pollard, Ph.D., deputy director of the Albert Einstein Comprehensive Cancer Center in New York.
The pictures, achieved through the convergence of multi-photon laser microscopy and the generation of transgenic mice with fluorescent tumors, are providing some of the first visual, real-time evidence linking cancer to inflammation in living animals, says Condeelis, a biophysicist who directs Einstein’s Analytical Imaging Facility.
Many questions remain, but new technologies and a flood of research studies are redefining the old, relatively static model of cancer. Tumors are astonishingly dynamic and versatile. They depend—in absolutely crucial ways—on interactions with surrounding normal tissue. This new view of cancer is shattering old assumptions, while at the same time raising hopes for earlier diagnosis, better treatment and, perhaps most provocatively, prevention.
“Prevention is going to become the dominant approach to cancer within the next 20 to 30 years… just as it has for cardiovascular disease,” predicts Ernest Hawk, M.D., MPH, vice president for cancer prevention and population sciences at the University of Texas M.D. Anderson Cancer Center in Houston. “That will come about through an improved understanding of the various pathways… that influence inflammation… And that’s where it really starts to get exciting.”
For nearly a century and a half, scientists have suspected that many cancers are caused by inflammation—the complicated process by which the body heals wounds and fights off invading pathogens.
Prime examples: colorectal cancer in patients with inflammatory bowel disease; lung cancer that follows chronic inflammation from inhaled asbestos particles; and tumors of the stomach and liver—among the most common cancers worldwide—that are linked to pathogen-induced inflammation. Inflammation also may contribute to the development of prostate cancer, which, next to lung cancer is the second leading cancer killer in American men.
Another link in this causal chain was forged about 10 years ago, when population-based studies detected a 40 percent to 50 percent drop in the relative risk of developing colorectal cancer among people who regularly use aspirin.
The finding came at a fortuitous moment: during the previous two decades, scientists had learned that aspirin worked by blocking production of prostaglandins, ubiquitous lipid-signaling molecules that are involved in a host of physiological processes. Two prostaglandin-generating enzymes, the cyclooxygenases, had been identified, and one of them—COX-2—was known to play an important role in pain and inflammation.
In 1994, Raymond N. DuBois, M.D., Ph.D., and his colleagues at Vanderbilt University Medical Center found high levels of COX-2 in cancerous colon tissue. Three years later, they had stopped human colon cancer cells from growing in the laboratory by blocking the COX-2 enzyme.
By this time, Celebrex—the first selective COX-2 inhibitor—was on its way to market for treatment of rheumatoid arthritis. Researchers began testing the drug in patients with familial adenomatous polyposis (FAP), an inherited condition characterized by the appearance of multiple polyps in the colon that nearly always become malignant. In June 2000, an international team of researchers, including DuBois and Hawk, reported that the use of Celebrex led to a significant reduction in the number of polyps in patients with FAP.
The landmark finding, published in The New England Journal of Medicine, opened the floodgates for studies aimed at preventing other cancers by blocking production of prostaglandins. Since then, DuBois and his colleagues have found that one of the members of the prostaglandin family, PGE2, seems to be specifically involved in spurring the proliferation and invasive potential of colorectal cancer cells, and it may do so—at least in part—through the receptor for epidermal growth factor (EGF), which stimulates cell growth.
“This is what I’m really excited about,” says DuBois, since 2007 provost and executive vice president for academic affairs at the University of Texas M. D. Anderson Cancer Center. “It just continues to support our whole concept from 1994, that (COX-2) is really playing a role” in colon cancer.
COX-2 is not the only performer in this drama. During the past half-century, scientists have chronicled a burgeoning list of growth factors, cytokines, genetic switches and protein-chomping enzymes that play interacting—and overlapping—roles in inflammation and cancer.
These chemicals are generated in response to emergencies such as injuries and infection by a variety of cell types and through a variety of “signaling pathways,” including activation of the COX-2 enzyme and the resulting production of prostaglandins. They include:
- Growth factors that trigger cell proliferation, and which block signals that otherwise would lead to cell death, a process known as apoptosis;
- Enzymes that repair and “remodel” tissue after an injury, and which spur the growth of new blood vessels to feed it; and
- Immune regulators, including cytokines, that call in the reinforcements of immune and inflammatory cells to fight infection and help heal wounds, and others that “call off the troops” to limit collateral damage to surrounding healthy tissue.
These pathways exist in exquisite balance. When the balance is tipped, or when certain genetic switches are turned on, certain growth factors and cytokines may be “over-produced” while others are suppressed. The result can be chronic inflammation, damage to tissues and abnormal growth. A growing number of scientists believe, therefore, that understanding the unique molecular and cellular “micro-environment” in which the tumor thrives is key to improving cancer therapy and prevention.
This is not an easy puzzle to solve. The “imbalance” of growth factors and cytokines that promotes cancer may differ depending on where in the body the tumor began and its stage of growth. The role of inflammation also may differ depending upon the circumstance.
In some cases, chronic inflammation may trigger tumor growth. Inflammatory cells, including mast cells, neutrophils and macrophages, can generate highly reactive molecules of oxygen and nitrogen that can literally punch holes in bacteria. Too much of this firepower, however, can damage the DNA of nearby cells, leading to out-of-control growth. Limiting inflammation in these cases could be an effective way to prevent the development of cancer.
Mutations in other cases may result from exposure to irradiation, carcinogenic chemicals or viral proteins. In this case, inflammation may be a “promoting” rather than initiating event. There is increasing evidence that the tumor can attract inflammatory cells, and use their growth-promoting factors to help it grow and spread. In this sense, the tumor “hijacks” the normal functions of inflammation for its own ends.
There is some evidence that tumor cells send out chemical signals—called chemokines—that attract inflammatory cells. According to one model, macrophages literally roll along the lining of blood vessels, following a trail of increasingly concentrated chemokines, like bloodhounds tracking a scent.
When the macrophages reach the site of the tumor, they squeeze through the blood vessel lining into the underlying tissue. Normally, they would sound the alarm, calling other elements of the immune system to attack the tumor. Instead, they are “re-educated” by the tumor to produce a variety of factors that nourish the tumor and enable it to break through the connective tissue that restrains it.
These “tumor-associated macrophages,” as they are called, also can release immunosuppressive factors—various members of the interleukin family of cytokines—that blunt the ability of the body’s immune surveillance system to detect and attack the tumor. So can other white blood cells (lymphocytes) that infiltrate tumors.
One possible way to stop this process and prevent tumor progression is to block expression of chemokines that chronically recruit inflammatory cells to the site where the tumor is developing, suggests Ann Richmond, Ph.D., Ingram Professor of Cancer Research at Vanderbilt. Richmond is co-discoverer of one of the first chemokines that has been shown to affect melanoma tumor growth.
In 2001, Pollard and his colleagues at the Albert Einstein Cancer Center reported that colony stimulating factor-1 (CSF-1), a major macrophage growth factor and chemokine, seemed to be essential in a mouse model of breast cancer for “metastasis,” spread of a tumor from its site of origin to other parts of the body.
Mice that lacked the gene for CSF-1 still developed mammary tumors, but the spread of the metastatic tumors to the lungs was significantly delayed. In addition, these tumors did not have the normal infiltration of macrophages seen in mice with the normal CSF-1 gene. When the gene was reintroduced into the mice, metastasis—in conjunction with macrophage infiltration—was restored. “At least in this model,” Pollard explains, “macrophages are bad news.”
Macrophages are not the only cell type that can fan the inflammatory flames of cancer. Zena Werb, Ph.D., Lisa Coussens, Ph.D., and their colleagues at the University of California, San Francisco have found that cells in the connective tissue surrounding the tumor also can send out signals that help it grow and spread. The cells “talk” to each other, just as they do during development of mammary glands or hair follicles. “The environmental side and the tumor side co-evolve,” explains Werb, professor and vice chair of Anatomy at UCSF.
Most cancers arise from the epithelium, the layer of cells that separate underlying tissues from the outside world. The epidermis of the skin, the linings of the lungs and digestive tract—all are examples of epithelial tissues. Underneath is the stroma, which provides the connective tissue, blood vessels, nerves and other vital physiological functions. Between cells is the extracellular matrix, which plays a role in tissue development and healing. It is here that the cells travel on their fibrous “superhighways” from one location to another.
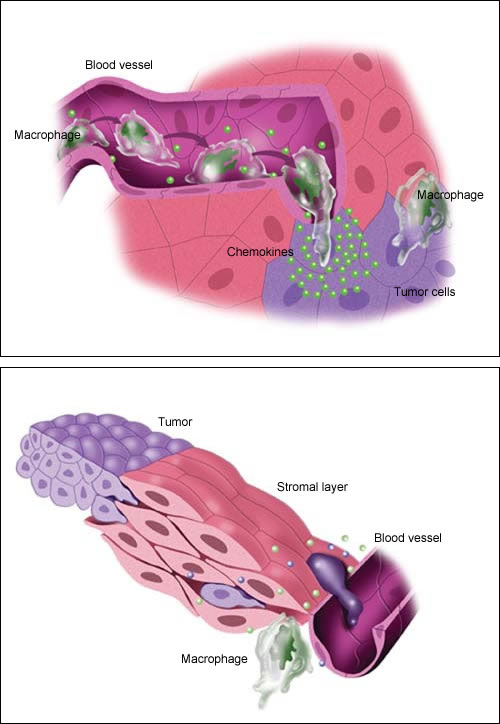
According to one model, macrophages roll along the lining of blood vessels, “tracking the scent” of chemical messengers called chemokines released by tumor cells. Once at the tumor site, they are “re-educated” by the tumor to produce factors that help it grow and spread.
Guided by factors in their “micro-environment,” some tumor cells find their way to a blood vessel. Once in the bloodstream, a chemokine trail—or possibly a blood cell “chaperone”—leads them to another tissue. This process is known as metastasis.
An example of this cellular “cross-talk” is the tissue remodeling that occurs during development or wound healing with the help of the matrix metalloproteinases (MMPs), a family of enzymes involved in a wide range of physiological processes. “Not only do they degrade the matrix, they release biologically active factors that are involved in calling in inflammatory cells and in angiogenesis,” says Lynn Matrisian, Ph.D., chair of Cancer Biology at Vanderbilt who, as a post-doctoral fellow, cloned the first full-length MMP in the mid-1980s.
MMPs also are believed to contribute to metastasis, the major cause of death from cancer, by helping to increase the tumor’s blood supply and means of escape to other parts of the body. The first synthetic MMP inhibitor was tested in humans in 1992, but by 2002, several clinical trials had failed to show any survival benefit.
That’s not surprising, says Matrisian, past president of the American Association of Cancer Research. MMP inhibitors “don’t stop cancer in its tracks,” she says. “What we learned is they change the rate of progression (of the disease).”
In 2002, Matrisian, Coussens and their Vanderbilt colleague Barbara Fingleton, Ph.D., predicted in an article in Science magazine that MMP inhibition would help prolong survival to the point that patients died of “old age” before they died of cancer. “But you can’t give it at the ninth hour,” Matrisian says. “You have to give it earlier, or you have to give it in combination (with other drugs)… or you have to think about prevention.”
Research is continuing. Matrisian and colleagues around the world are developing biomarkers and optical imaging techniques to determine how effectively the inhibitors block the enzymes, and new animal models that more closely resemble the human condition.
Other researchers are tackling metastasis from the point of view of the errant tumor cell that makes it into the bloodstream. In patients with cancer, “there are (an estimated) million or so tumor cells circulating at any one time, and yet only one or two of those end up as metastases,” Pollard says. “Does that mean that (only) one in a million cells… is able to metastasize, or that there are a million cells able to metastasize but only a one-in-a-million chance of them lodging in the right place?”
One theory is that once out in the bloodstream or lymphatic system, the tumor cell tracks a “trail” of chemokines to a specific tissue that is producing the signal—essentially reversing the course that inflammatory cells took to get to the primary tumor. “Another possibility which is a bit… more outlandish,” he says, “is that tumor cells are ‘chaperoned’ by (blood) cells.” In either case, the spread of cancer may be guided by specific chemokines released by various tissues.
Pollard hopes the new imaging technology the Einstein group has developed will help solve the mysteries of metastasis. “We should be able to follow cells as they move around the body in ways that we’ve not been able to do before,” he says.
Ultimately, the control of cancer may depend on a better understanding of the complex chemical signals that guide these cells. “These are the big questions,” says DuBois. “How do these inflammatory cells potentiate tumor development at the molecular level? What is the precise role of the microenvironment, and what regulates the inflammatory cell recruitment into the neoplastic (tumor) microenvironment? We don’t have answers to that.”
With the help of techniques such as DNA microarray, scientists are identifying genes that play a role in inflammation and cancer. They’re studying what happens when the genes are “knocked out” in mouse models. One goal is to learn how to “manipulate” macrophages, for example, by altering the balance of cytokines in a way that would turn them from the tumor’s friend to a foe.
On the clinical side, researchers are trying to identify “bio-markers,” or patterns of proteins in blood samples from patients that can be correlated with types and stages of cancer, and their response to treatment. Another approach seeks to identify groups of patients with genetic differences, called polymorphisms, who would be most likely to benefit from targeted cancer therapies.
With these approaches, “you will have determined on some level what genetic pathways or patterns of protein expression are altered in the patient’s tissue,” Richmond explains. “Then you might be able to predict which regime to use for that patient by targeting the defect in that specific patient’s tumor.”
For example, if over-production of pro-inflammatory cytokines and chemokines can be detected, and if drugs that specifically target these signaling molecules or their receptors can be given to block this step in tumor progression and metastasis, “we will likely increase our success with both prevention and therapeutic intervention in cancer,” she says.
What’s needed, says DuBois, is a unifying theme. “There are different components of the inflammatory response that play a role in cancer,” he says. “We’ve been looking at them individually. Hopefully people can go back and look at this, and try to see if they can make sense of some of the research that’s been done.”