by Paul Govern
In a study out this week in Science Translational Medicine, an international team led by researchers at Vanderbilt University Medical Center sheds new light on the cause of pulmonary fibrosis and demonstrates a way to impede the disease in mice.
The study results imply that certain drugs already at market might be able to be repurposed to inhibit or halt this major respiratory disease.
“I’m very excited by the implications of our study for drug repurposing and the important benefits that might accrue for a great many patients with lung disease,” said Wonder Drake, MD, professor of Medicine and Pathology, Microbiology and Immunology, who led the study with Lindsay Celada, PhD, research instructor in Medicine.
“I’m doubly excited at the prospect of these clinical trials proceeding right here at VUMC. A combination of clinical investigative talent and research infrastructure makes VUMC the natural place to advance our lab’s findings into the clinical realm.”
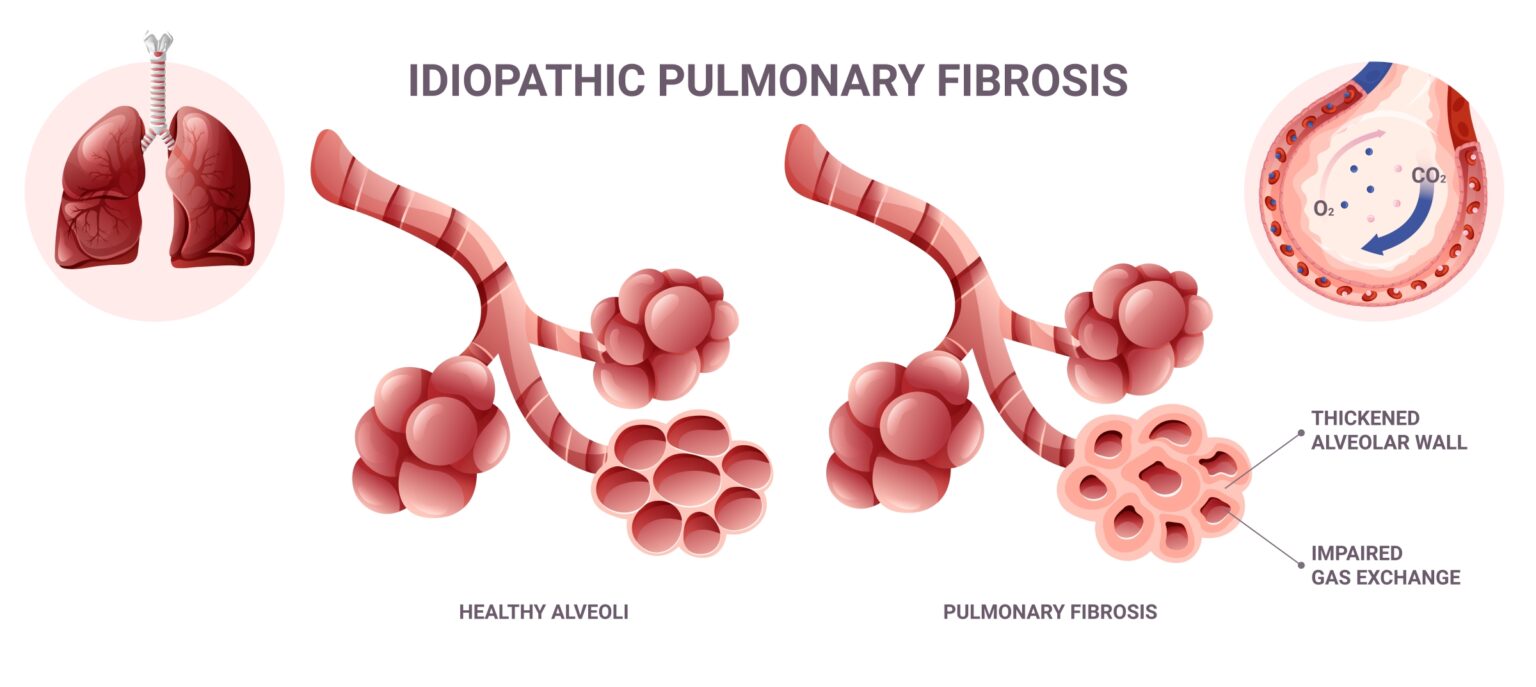
People with pulmonary fibrosis are perpetually short of breath. Connective tissue in their lungs has become thickened or scarred, reducing the supply of oxygen to the blood.
Fibrosis occurs in a number of diseases affecting the lungs, among them sarcoidosis and idiopathic pulmonary fibrosis (IPF). Median survival with IPF is approximately three years. Pulmonary fibrosis can also occur in response to bleomycin, a drug in use since the 1960s to treat cancer.
Pursuing cues from an assortment of previous findings, the new study uncovers a common immunological mechanism underlying these three distinct disease types.
In both IPF and bleomycin-induced fibrosis, previous studies had indicated connections with a pro-inflammatory cell signaling protein called interleukin-17 (IL-17A). Meanwhile, people studying sarcoidosis had found dysfunction in a type of white blood cell, the CD4+ T cell.
Drake had traced this dysfunction to upregulation of PD-1, or programmed cell death protein 1, a cell surface receptor that inhibits autoimmunity.
Fibroblasts are cells that produce the structural framework of animal tissues, including wound-healing tissues. IPF in particular has been shown to involve overactivation of lung fibroblasts.
The team placed normal lung fibroblasts in culture with CD4+ T cells harvested from people with IPF or sarcoidosis. Type 1 collagen production, a hallmark of fibrosis, results in both these cultures; but not so in the case of CD4+ T cells from patients with healthy lungs.
Employing an array of techniques and covering a lot of ground, the study introduces a causal story that starts with PD-1 upregulated CD4+ T cells, leading to increased expression of a gene transcription activator called STAT3, and then on to increased production of IL-17A, resulting in fibrosis.
“Though they haven’t typically been considered important in pulmonary fibrosis, we’re showing a central role in a range of diseases for CD4+ T cells with PD-1 upregulation,” Celada said.
With its wide scope, the study brings attention to three potential drug targets for which drugs are already available: blocking PD-1 can significantly reduce fibrosis in mice, the team shows; using human cells in vitro, they show that chemical inhibition of STAT3 reduces components of fibrosis, such as collagen production; and finally, the study brings added attention to the role of IL-17A in sarcoidosis pulmonary fibrosis.
The study was supported by grants from the National Institutes of Health (HL117074, HL127301).